
Zurück zu HOME
EINE NÜTZLICHE MUTATION: SICHELZELLENANÄMIE?
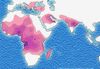 Bild 138: Wikimedia, Autor Muntuwandi, Lizenz: Creative Commons Attribution-Share Alike 3.0
Bild 138: Wikimedia, Autor Muntuwandi, Lizenz: Creative Commons Attribution-Share Alike 3.0Als Beispiel einer nützlichen Mutation wird oft die in Afrika auftretende Sichelzellenanämie erwähnt. Diese Krankheit verhindert einen Malarieabefall.
Die Tatsache an sich ist Richtig, allerdings um einen hohen Preis. Hier ist ein Artikel über die Krankheit der Sichelzellenanämie. Er ist sehr mit Fachausdrücken gespickt, daher gebe ich eine kurze Zusammenfassung:
Die Blutkrankheit der Sichelzellenanämie betrifft die roten Blutkörperchen. Diese Mutation führt u.a. zu Durchblutungsstörungen die lebensbedrohliche Ausmaße annehmen kann. Aufgrund dieser Mutation kann es jedoch bei der leichteren Form zu keiner Malariaerkrankung kommen.
Es wäre in etwa so: Ich verliere meinen rechten Arm, folglich kann ich an dieser Seite keinen Tennisarm mehr bekommen.
Der Verlust ist größer als der Gewinn. Hier ist der Artikel:
Die Sichelzellenanämie (med.: Drepanozytose), auch Sichelzellanämie (englisch: sickle cell anemia) ist eine erbliche Erkrankung der roten Blutkörperchen (Erythrozyten). Sie gehört zur Gruppe der Hämoglobinapathien (Störungen des Hämoglobins). Bei den Betroffenen liegt eine Mutation der beta-Kette des Hämoglobins vor. Es können entweder alle beta-Ketten betroffen sein (schwere, homozygote Form) oder nur ein Teil. (mildere, heterozygote Form).
 Bild 139: Gemeinfrei
Bild 139: GemeinfreiDie Betroffenen bilden ein abnormes Hämoglobin (Sichelzell-Hämoglobin, HbS), das bei Sauerstoffmangel zur Auskristallisation neigt. Dabei verformen sich die roten Blutzellen zu sichelförmigen Gebilden und verstopfen kleine Blutgefäße. Dadurch kann es bei der homozygoten Form zu anfallartigen schmerzhaften, z. T. lebensbedrohlichen Durchblutungsstörungen (Sichelzellkrisen) kommen. Heterozygot Betroffene, bei denen nur eines der beiden Hämoglobin-Gene verändert ist, sind vor den schweren Verlaufsformen der Malaria geschützt. Dadurch ist das mutierte Hämoglobin-Gen in Malariagebieten relativ verbreitet.
Die Zerstörung roter Blutkörperchen führt zu einer schweren chronischen Blutarmut (hämolyische Anämie). Aufgrund der Neigung des Hämoglobin S zur Polymerisation und der sichelförmigen Deformierung der Erythrozyten kommt es zu Verschlüssen kleiner Arterien mit rezidivierenden Durchblutungsstörungen. Dies führt zu starken Schmerzen und Schäden in multiplen Organsystemen: Gehirn (ischämischer Schlaganfall), Milzinfarkt, Lunge (Lungenentzündung, pulmonale Hypertonie), Auge, Herz- und Nierenversagen, Muskeln, Knochen (Osteonekrose) oder Priapismus. Die Lebenserwartung ist vermindert.
Eine Glomerulopathie mit Hyperfiltration tritt bei bis zu einem Drittel der Patienten mit homozygotem Phänotyp schon in der Kindheit auf. Schäden im Bereich des Nierenmark führen zu Papillennekrosen, Verlust der Konzentrationsfähigkeit der Nieren und blutigem Urin (Makrohämaturie). Schäden im Bereich der Nierenkörperchen (Glomeruli) führen zu vermehrter Eiweißausscheidung im Urin (Mikro- und Makroalbuminurie, nephrotisches Syndrom). Bei der feingeweblichen Untersuchung ist die vorherrschende glomeruläre Schädigung die fokal segmentale Glomerulosklerose. Bei bis zu einem Drittel der Patienten kommt es in den ersten Lebensjahrzehnten zum Auftreten einer Proteinurie, in fünf Prozent zu einem terminalen Nierenversagen.[1][2]
Nur homozygote Träger des Sichelzellgens zeigen diese starke Ausprägung der Krankheit, bei der das gesamte Hämoglobin abnormes Sichelzellhämoglobin (irreguläres Hämoglobin) ist. In heterozygoten Trägern ist nur etwa ein Prozent aller Erythrozyten deformiert. Die Symptome verschlimmern sich erheblich, wenn die Patienten körperlich stark aktiv sind oder sich in großen Höhen befinden. Dies liegt daran, dass sich die Sichelform der Erythrozyten bei niedrigem Sauerstoffpartialdruck bildet, weil unter diesen Bedingungen das Hämoglobin faserig ausfällt (die Löslichkeit von Hämoglobin bei Sichelzellanämie ist 25-fach geringer als die Löslichkeit von normalem Hämoglobin).
Etwa ab dem sechsten Lebensmonat, wenn der Abbau des fetalen Hämoglobins bereits weit fortgeschritten ist, können erstmals Symptome auftreten. Sie äußern sich dann meist in einer sogenannten Sichelzellkrise: durch äußere Einflüsse, wie z. B. Anstrengung, sinkt der Sauerstoffpartialdruck im Blut, die Sichelzellen werden hämolytische.
- ↑ Harrisons Innere Medizin, 15. Auflage, S 1754
- ↑ Raimund Hirschberg: Glomerular hyperfiltration in sickle cell disease. In: Clinical Journal of the American Society of Nephrology: CJASN. 5, Nr. 5, 2010, S. 748-749. doi:10.2215/CJN.01340210. Abgerufen am 21. August 2010.
Ende des Artikels
Ob es sich hiermit um eine nützliche Mutation handelt sei dahingestellt.
Zurück zu HOME